help.ielm
The aim of iELM is to facilitate the exploration of linear motif interactions. iELM identifies motif-mediated interactions and their molecular interfaces both at sequence level and structural level.
Go to section explaining scoresGo to section explained Proteomic iELM
Go to Frequently asked Questions
Go to section explaining difference between iELM and Pfam HMMs
protein ielm
This section searches a database of pre-calculated data for SLiM-mediated interactions. Running the iELM method against the high-quality interaction data from the STRING resource produced this data
Input:
Any ID format from a dropdown menuOutput:
The output is divided into a tabular and graphical output: - The tabular output consists of the two tables: the first table (if
applicable) consists of results of SLiMs found within the query protein;
the second table (if applicable) consists of results of SLiM-binding
domains found within the query protein. It describes the two proteins (linked to UniProt) involved in
the SLiM-mediated interaction, the ELM functional class (linked to ELM)
and Pfam domain (linked to Pfam), as well as the position of the SLiMs
and SLiM-binding domains within the respective proteins along with a
score for each and an overall score for the entire interaction. The table also provides links to PepSite enabling structural
predictions for the interaction to be viewed. The table is fully
searchable and can be copied to clipboard or downloaded as a
comma-separated values (csv) document.
The tables have three parts: The left part contains
the motif UniProt ID, the motif type (ELM functional class), the location of the motif in the sequence and the associated scores. The central portion shows the UniProt ID of the motif-binding
domain, the domain type (Pfam), and the domain score (see below). The final part of the right gives
the total score for the prediction and the pepsite score where applicable.
- The graphical output consists of the modular architecture of the query protein extracted from the UniProt database along with graphical representations of the predicted SLiMs and SLiM-binding domains. This interface has tool tips describing the prediction and is clickable leading to the aforementioned tables to be filtered. The key is also clickable and filters the tables and figure based on the ELM functional categories.
Output Scores
The scores shown in the tables can be interpreted as followsIn all cases, the higher the score the greater the confidence.
Domain Score - similarity score compared to a known motif-binding domain with a higher score representing a greater confidence.
Motif Score - relative local conservation score. This compares the conservation of the amino acids in the motif as compared to those amino acids surrounding the motif. The higher the score the more trustworthy the results.
Disorder Score - This score is calculated by amino acid propensity using the IUPred prediction algorithm. Scores above 0.5 can be considered to be disordered, below 0.2 are likely to be ordered, between 0.2 and 0.5 could be either.
A cut-off system is used to reduce false hits and therefore only results with a disorder score above 0.3, a motif score above 0.6 and a domain score above 0.35 are displayed. Care should be taken in intepreting all scores close to and especially below these cut-off values.
View Structure
Please click on pepsite button to view prediction of structure (presently unavailable) For example results please press here
proteomic ielm
This section allows the user to input a protein-protein interaction network that is searched using the iELM method
Input:
Two inputs are allowed either a tabulated list of interactions or a list of IDs that will be searched in an all-against-all manner. Once again any ID format from a dropdown menu is allowed. There is a limit of 75,000 interactions for a tabulated list or 400 IDs for all-against-all search
Output
As with the iELM section the output is divided into two sections:- The tabular output is the same structure as described in ‘Run iELM’, accept only one table is displayed that contains all the interactions and the queried proteins are displayed to the left of the UniProt IDs. There is also an additional button called ‘Interaction’. Clicking this button leads to the production of a graphical representation of the protein-protein-interaction network linked to this interaction. An additional table displays those proteins associations not predicted to be SLiM-mediated.
- The graphical output contains the modular architecture as outlined above as well as a network of all the connecting interactions in one connection cluster of up to 75 proteins. The originally produced network is based on the best score but can be altered by pressing the ‘Interaction’ button. The edges of the network are coloured depending of the type of interactions and the nodes are coloured depending on whether they contain a SLiM, a SLiM-binding domain or both. The width of the edges reflects the score assigned by the method.
For example network please press here
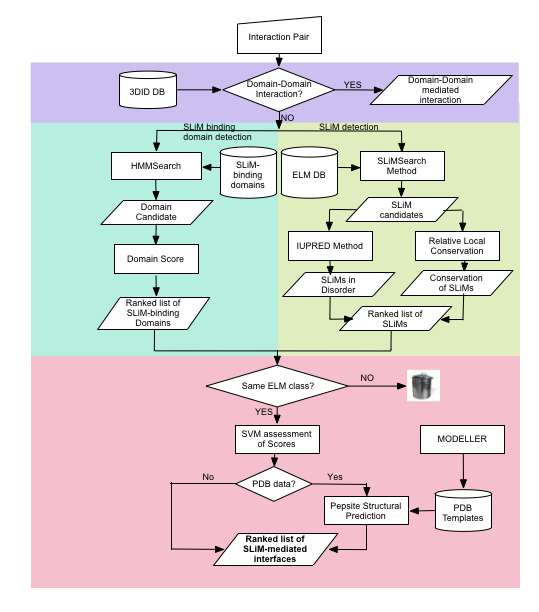
ielm algorithm schema
FAQ: frequently asked questions
1) Why are the graphics not working?
The graphics require JavaScript to be turned on. Please google the name of your browser and 'turning on JavaScript' to resolve this.
2) The proteomic button is not active.
Once again JavaScript is required, please see answer above.
3) I can not print, copy or get the csv files of the table?
These button require flash. please ensure flash is active.
Difference between iELM and Pfam HMMs
iELM HMMs are trained to recognise linear motif-binding domains based on annotation of known PDB structures and literature searches. This is updated regularly but poorly-studied domains may be missed. If so, Pfams HMMs can be used. These HMMs will identify all globular domains of a particular type but will not take into account the specificity linear motifs often have for sub-groups of a domain family.
EXAMPLE
When the human protein NHRF4_HUMAN is entered into the ‘run ielm’ search box with iELM HMMs, 3 LIG_PDZ_Class_1 binding domains are identified as well as one PDZ domain with no hits. The PDZ4 domain of NHRF4_HUMAN has apparently diverged too far from those domains known to bind LIG_PDZ_Class_1, so iELM cannot confidently identify this domain as being able to bind a LIG_PDZ_Class_1 motif. When searching iELM using Pfam HMMs all 4 PDZ domains are identified, however all three classes of PDZ motifs are found associated with each domain. NHRF4 PDZ domains are specific for LIG_PDZ_Class_1 motifs and therefore many of the hits for the other classes are likely to be false positive results. This information on specificity is incorporated into the iELM HMMs but not into the Pfam HMMS. In this way, iELM HMMs domains may be slightly too specific and miss true positives whereas Pfam HMMs are too unspecific and therefore identify a greater number of false positive hits.